- TECHNOLOGY
- DeepZema®
DeepZema®
Schematic representation of the components of DeepZema®
Drug discovery and development are a long and costly process. To bring a drug to market, biopharmaceutical companies typically spend more than $1 billion during the period of 10-15 years. In the traditional discovery setting, it may take 4-5 years to come up with just a drug candidate. Therefore, companies with technology that can help reducing the time and cost in discovery or development will lead the industry. Over the past few years, the world witnessed a sensational rise of AI-based drug discovery start-ups such as Insilico Medicine and Exscientia.
The common features of AI-based technologies at these companies appear to be (1) the omics-based identification of disease-related targets and (2) the multi-parameter lead optimization using generative models. While these approaches will certainly help the discovery process, they tend to demand some extent of financial and human resources that small start-ups cannot afford. Thus, Innovo Therapeutics adopted a more practical approach to address this issue by augmenting artificial intelligence to existing computational drug discovery technologies. These include ML-based target identification and ADME/Toxicity predictions. Instead of delving into reinforcement learning, we use traditional de novo design methods to generate new analogs which are subsequently submitted to docking. We then evaluate the binding poses with deep learning-based scoring functions and predict their ADME/Toxicity properties.
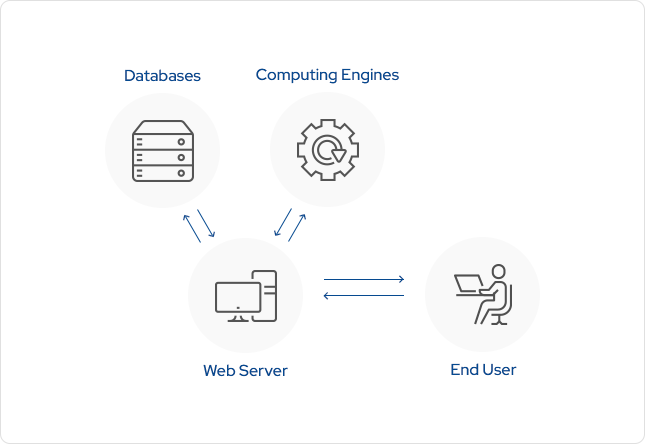
All these computational procedures were combined to form a proprietary computational platform called DeepZema®. DeepZema® has been developed with philosophies of (1) simplicity (2) democratism and (3) practicality. Thus DeepZema® has a simple web interface for easy access by experts and non-experts alike and returns the results in real time. DeepZema® consists of a typical server-client system with the server connected to databases and computing engines.
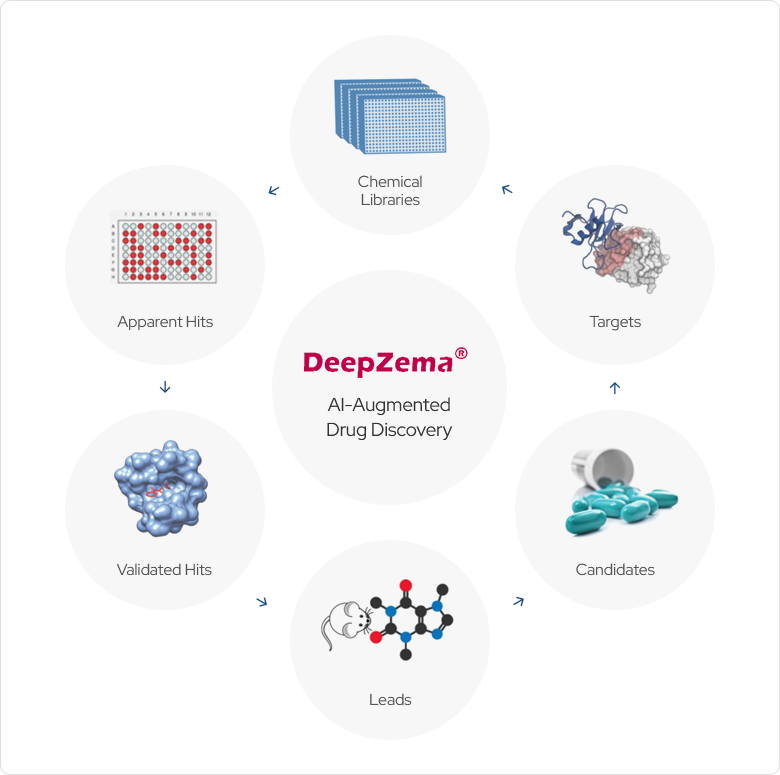
Application areas of DeepZema®
- • Ligand similarity and shape-based target identification.
- • Browsing/searching internal/external databases such as Dotmatics,
PubChem, ChEMBL, PDB, DrugBank and ChemBank. - • Virtual screening of chemical libraries and hit confirmation
(identification of false positives). - • Conformational analysis and 3D shape alignment of ligands.
- • Lead optimization (QSAR, de novo design, protein-ligand docking)
- • ML-based ADME/Toxicity calculations
- • Developability assessment (melting properties, pH dependence of solubility,
permeability, formulation, etc) - • Project management (current status report, etc)
Due to multiple functionalities of DeepZema®, it is not practical to explain all facets here.
Instead we will just give some examples to show how DeepZema® works.
- Kinome Profiling
- Target Prediction
- ADME/Toxicity Prediction

When we develop a kinase inhibitor drug, the selectivity profile over the whole kinome is very important in order to obtain a desired efficacy and level of safety. Thanks to the kinome profiling module implemented in DeepZema®, we can examine the selectivity profile of compounds before synthesis. For example, in the process of CDK7 inhibitor optimization at Syros pharma., both SY-5609 and SY-5102 showed a potent CDK7 activity but SY-5609 had a much better selectivity profile in the 485 kinase panel screen.
Thus SY-5609 has been chosen for the development candidate (J. Med. Chem. 2022, 1458). By using the DeepZema® kinome profiling, this situation can be easily reproduced as shown below. This means that DeepZema®’s kinome profiling tool can be used to guide a selectivity optimization during the discovery process.
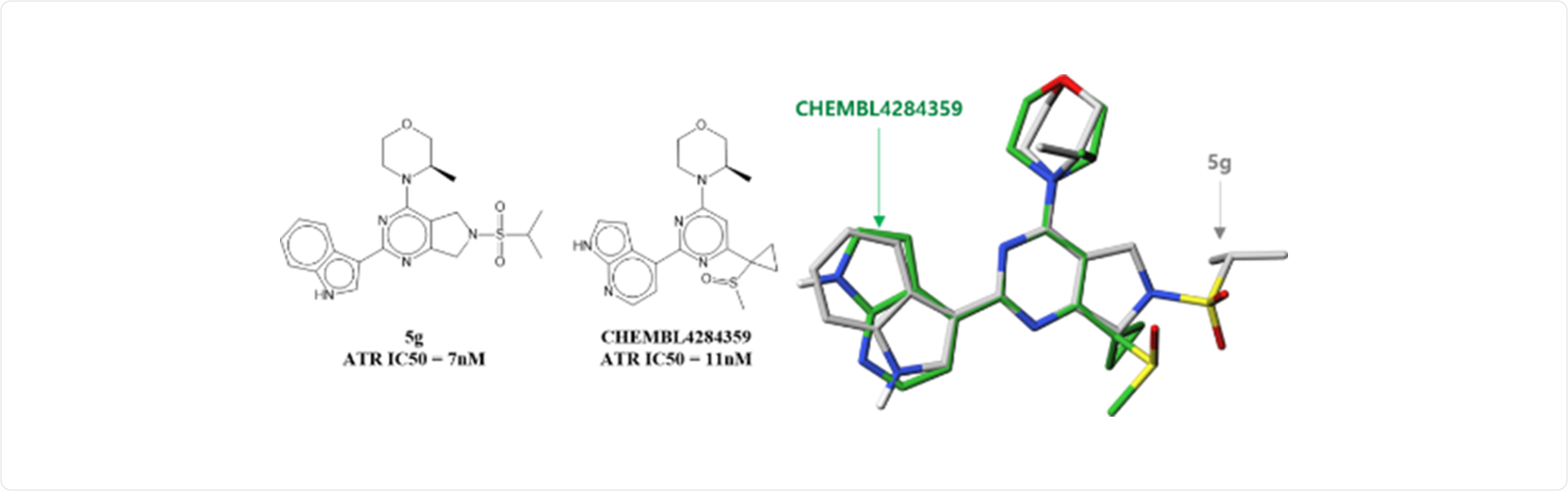
DeepZema® offers two different approaches for prediction of targets, i.e., conformal prediction and 3D shape matching methods. Compound 5g is an ATK kinase inhibitor with IC50 of 7nM (BMCL 2022, 128651). Conformal prediction on this compound gives mTOR and ATR as plausible targets. On the other hand, 3D shape alignment of 5g to known kinase inhibitor library resulted in a list of kinases such as mTOR, PI3K, CLK4, MAPK1 and ATR kinases. Target prediction may be used for identifying targets of phenotypic screening hits or for identifying off-targets of given compounds.
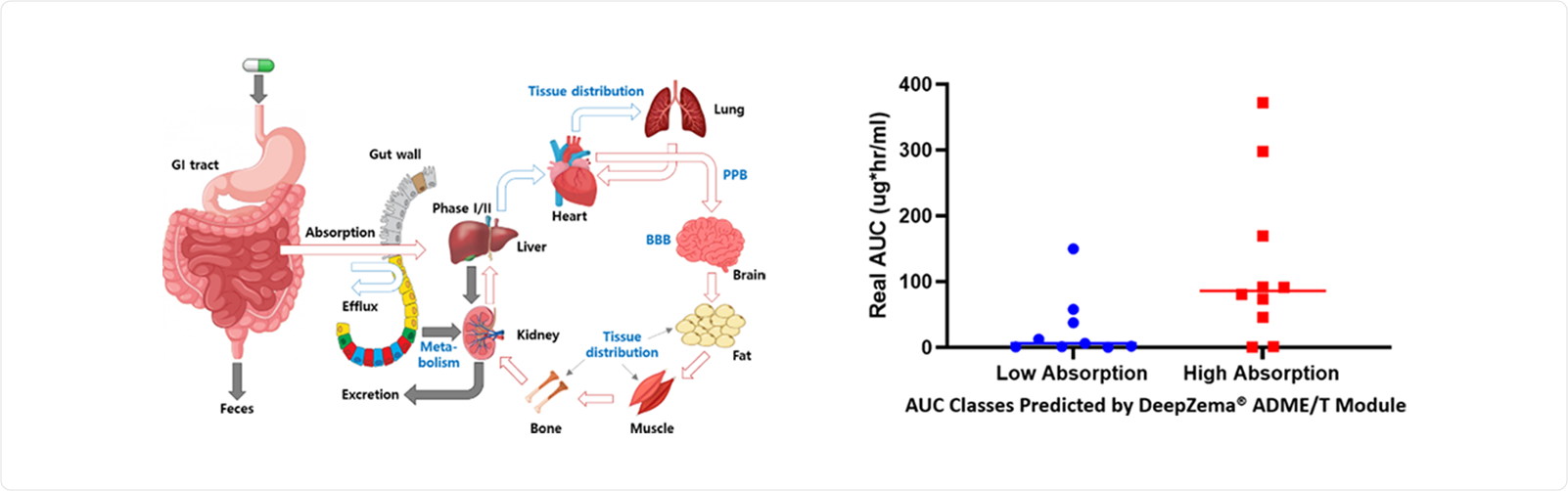
The following figure illustrates ADME for oral administration of drugs (Predicting ADME Properties of Chemicals, K. T. No et al, Springer 2017). The fate of orally administered drugs is determined by their ADME (Absorption, Distribution, Metabolism and Excretion) properties. These properties must be optimized for the drug to reach the target organ to take pharmacological effects. During the early stage of drug discovery, in silico methods can reduce the time and cost of ADME evaluation in the lead optimization process.
ML and deep learning-based models for a variety of ADME properties as well as toxicity endpoints are available in DeepZema®. For example, DeepZema® implements (1) absorption (oral BA, HIA, Pgp inhibition/substrate, MDCK permeability, skin penetration), (2) distribution (BBB penetration, PPB), (3) metabolism (Cyp inhibition) and (4) Excretion (Clint, metabolic stability, half life).
We had experimental AUC values for 19 proprietary compounds. These experiments were carried out by an independent third-party laboratory. We also calculated low/high AUC categories for these compounds using DeepZema® and compared the calculated values with experiments.